Cystic fibrosis isn't just another lung disease. It’s a genetic condition that changes how your body handles salt and water at the cellular level, turning mucus from a slippery protector into a thick, sticky trap. This isn’t something that happens over time-it’s built into your DNA from birth. And while it’s been known since the 1930s, the last decade has rewritten the story of what living with cystic fibrosis can look like.
What Actually Goes Wrong in Cystic Fibrosis?
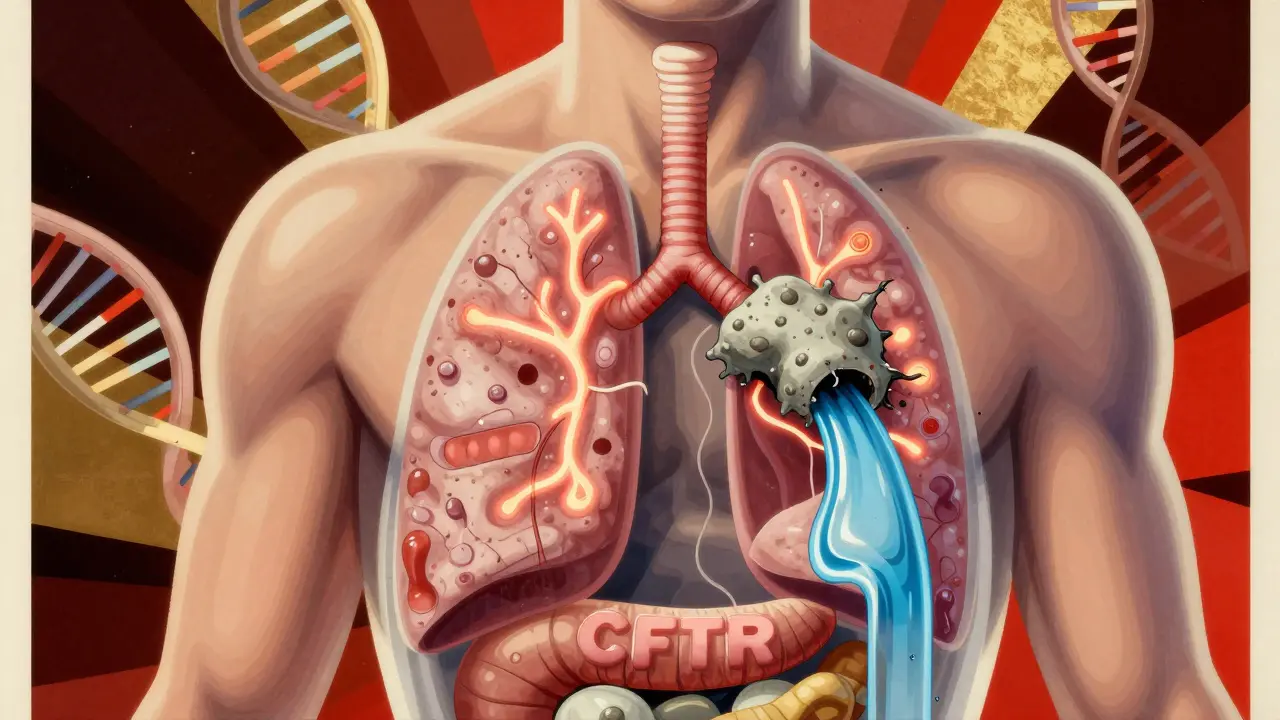
At the heart of cystic fibrosis is a single gene: CFTR. When this gene works right, it acts like a gatekeeper on the surface of cells in your lungs, pancreas, liver, and sweat glands, letting chloride ions move in and out to keep fluids balanced. But if you inherit two broken copies-one from each parent-that gate gets stuck. No chloride flow means no proper hydration of mucus. Instead of thin and slippery, it becomes thick, sticky, and impossible to clear.
This isn’t just a lung problem. That thick mucus clogs the pancreas, stopping digestive enzymes from reaching your intestines. About 85% of people with CF need to take pancreatic enzyme capsules with every meal just to absorb nutrients. It blocks bile ducts in the liver, causes infertility in nearly all men, and leads to dangerously high salt levels in sweat-the very test doctors use to diagnose it.
The most common mutation, F508del, shows up in about 70% of cases worldwide. But there are over 2,000 known CFTR mutations, and not all respond the same way. That’s why treatment can’t be one-size-fits-all.
Why CF Is Different From Other Respiratory Diseases
People often confuse cystic fibrosis with conditions like asthma or chronic bronchitis. But those are usually triggered by environment or lifestyle. CF is genetic, lifelong, and multi-system. Even compared to Primary Ciliary Dyskinesia (PCD)-another rare disease that causes mucus buildup-CF is fundamentally different. PCD breaks the tiny hair-like structures (cilia) that sweep mucus out of the lungs. CF doesn’t break the broom; it turns the dirt into concrete.
This distinction matters because it shaped the treatment revolution. While PCD still has no targeted drugs, CF became the first genetic disease where scientists could design medicines to fix the broken protein itself. That’s precision medicine in action.
The Game-Changers: CFTR Modulators
Before 2012, treatment for CF was about managing symptoms: chest physiotherapy, antibiotics, inhalers, enzymes. Survival into adulthood was rare. The median life expectancy in 1960 was just 14 years.
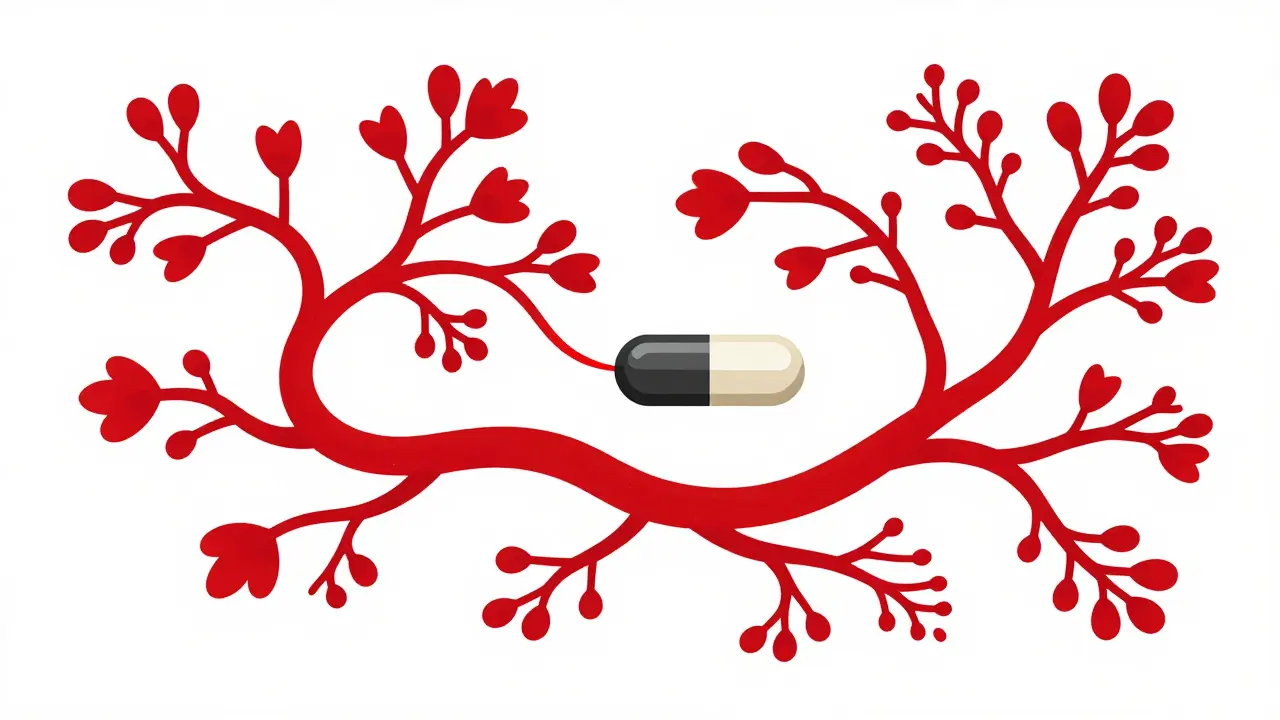
Then came ivacaftor (Kalydeco), the first CFTR modulator approved in 2012. It worked for people with the rare G551D mutation, boosting lung function by over 10% in clinical trials. But the real breakthrough came with triple therapy: elexacaftor/tezacaftor/ivacaftor (Trikafta), approved in 2019. For people with at least one F508del mutation-which covers about 90% of patients-it improved lung function by 13.8% and cut pulmonary exacerbations by 63%.
Today, 90% of people with CF in the U.S. have access to at least one modulator. Life expectancy has jumped to over 50 years. More than half of the CF population is now adult. That’s not just progress-it’s a transformation.

Who Still Doesn’t Benefit?
But here’s the hard truth: about 10% of people with CF have mutations that don’t respond to current modulators. These are often Class I mutations-nonsense mutations where the body stops making the CFTR protein altogether. For them, modulators offer nothing. Their daily routines haven’t changed: hours of airway clearance, multiple inhalers, enzyme capsules, and constant vigilance against infection.
And even for those who do respond, it’s not perfect. Side effects like elevated liver enzymes happen in about 3% of users. Some report severe fatigue, headaches, or mood changes. One patient on a CF support forum described stopping Trikafta after three months because their liver enzymes spiked to dangerous levels. For others, the cost is unbearable. In the U.S., these drugs cost around $300,000 a year. Even with insurance, out-of-pocket costs average $1,200 a month. That’s why only 35% of the global CF population has access to these therapies.
What Daily Life Looks Like Now
Before modulators, adults with CF spent 2 to 3 hours a day on treatments: chest percussion, vibrating vests, nebulizers, enzymes, vitamins. Many missed work or school. Social life revolved around avoiding germs.
Now, many report a dramatic shift. One 28-year-old with two F508del mutations said Trikafta cut their daily airway clearance from 90 minutes to 20. They gained 15 pounds in six months. They started hiking. They got a job. They didn’t need to cancel plans because they were sick.
A 2022 survey of nearly 8,000 CF patients found that 89% felt better breathing, 76% had fewer hospital visits, and 68% gained weight. But 42% still struggled with financial stress. The treatment burden hasn’t vanished-it’s just changed.

The Future: What’s Coming Next?
The Cystic Fibrosis Foundation is pouring money into closing the gap for the 10% left behind. Fifteen clinical trials are active as of 2025, including:
- mRNA therapies to trick cells into making functional CFTR protein
- CRISPR gene editing to fix mutations directly in DNA
- New inhaled antibiotics designed to break through stubborn Pseudomonas biofilms
Trikafta was recently approved for kids as young as two. That means early intervention is now possible before lung damage sets in. And with Vertex Pharmaceuticals holding 95% of the modulator market, pressure is building to lower prices and expand access globally.
The World Health Organization has called CF a stark example of global health inequity. While 85% of eligible U.S. patients get modulators, fewer than 10% do in low-income countries. Most CF-related deaths today happen in places where these drugs are unaffordable or unavailable.
What This Means for Patients and Families
If you or someone you love has CF, the message is clear: hope is real. The science is working. Life expectancy is no longer a death sentence. But it’s not magic. It’s hard work-medication schedules, clinics, insurance battles, side effects, and emotional tolls.
For families with newborns diagnosed through screening, the path is different now. Instead of preparing for early loss, they’re planning for college, careers, travel. The disease hasn’t disappeared, but its grip has loosened.
For those without modulator options, the fight continues. Research is accelerating. Community support is stronger than ever. And the goal-equitable access to life-changing therapy-is no longer a dream. It’s a demand.
Cystic fibrosis is no longer just a pediatric disease. It’s a chronic condition with a future. And that future is being written right now-in labs, clinics, and living rooms around the world.
Is cystic fibrosis contagious?
No, cystic fibrosis is not contagious. It’s a genetic disorder passed down from parents to children through inherited mutations in the CFTR gene. You can’t catch it from someone else like a cold or the flu. Only people who inherit two faulty copies-one from each parent-will develop the disease.
Can you outgrow cystic fibrosis?
No, you cannot outgrow cystic fibrosis. It’s a lifelong condition caused by your DNA. However, new treatments like CFTR modulators have dramatically improved life expectancy and quality of life. Many people with CF now live into their 50s and beyond, managing the disease as a chronic condition rather than a terminal one.
How do CFTR modulators work?
CFTR modulators are drugs that fix the faulty CFTR protein at the cellular level. Some help the protein reach the cell surface (correctors like tezacaftor), others help it function better once it’s there (potentiators like ivacaftor). Triple therapy combines both types to restore chloride flow, thin mucus, and reduce infections. They don’t cure CF, but they treat the root cause-not just the symptoms.
Why are CFTR modulators so expensive?
CFTR modulators are expensive because they’re highly specialized drugs developed for a rare disease. Developing them took decades, billions in research funding, and complex clinical trials. Vertex Pharmaceuticals, the main manufacturer, holds patents and market exclusivity. While the Cystic Fibrosis Foundation helped fund early research, the final drug pricing reflects pharmaceutical industry models for orphan drugs-often exceeding $300,000 per patient per year in the U.S.
What’s the life expectancy for someone with CF today?
As of 2022, the median predicted survival for someone with cystic fibrosis in the U.S. is 50.9 years. That’s up from just 14 years in 1960. For children born today with access to modulators, many are expected to live well into their 60s or beyond. Survival varies based on mutation type, access to care, and overall health-but the trend is clearly upward.
Can people with CF have children?
Most women with CF can become pregnant and carry a child to term, though they may need extra medical support. For men, nearly all (97-98%) are infertile due to missing or blocked vas deferens. However, assisted reproductive technologies like IVF with sperm extraction can allow biological fatherhood. Genetic counseling is strongly recommended before planning a pregnancy, since CF is inherited.




Cara C
December 21, 2025 AT 16:26Just read this whole thing and honestly? I’m in tears. Not because it’s sad, but because for the first time, I can imagine my niece having a full life. She’s 5. Started Trikafta last year. She’s running around like a wild thing now. No more 2-hour nebulizer marathons before bed. It’s not perfect, but it’s hope made real.
Thank you for writing this.
Michael Ochieng
December 22, 2025 AT 04:06As someone from Kenya who’s worked with CF families in Lagos and Nairobi, I can tell you this: the gap isn’t just about money-it’s about awareness. Most clinics don’t even test for CF because they assume it’s a ‘white person disease.’ We’ve got kids dying from pneumonia that’s just CF going untreated. This isn’t a US problem. It’s a global failure.
And yeah, Trikafta’s expensive-but if we can fund a Mars rover, why can’t we fund a kid’s chance to breathe?
Grace Rehman
December 23, 2025 AT 09:10So let me get this straight-we spent decades building drugs that fix a broken gate… but only if you’re rich enough to pay for it?
And we call this progress?
Meanwhile, the people who invented the gate in the first place-the ones who passed down the broken gene-are still being told to ‘manage it’ with chest thumping and enzyme pills while billionaires buy islands.
It’s not science that’s failing here. It’s capitalism. And it’s ugly.
Also side note: anyone else notice how every single article about CF modulators says ‘hope’ but never says ‘who gets to have it’? Just saying.
Jerry Peterson
December 24, 2025 AT 09:16My cousin’s kid was diagnosed at birth. We thought it was over. But now? She’s in 4th grade, plays soccer, doesn’t miss school. I remember when the first modulator came out-everyone was excited, but nobody knew if it’d work for her mutation. Turns out, she’s F508del. Trikafta changed everything.
Still, I worry about what happens when she’s 30 and insurance drops her. This isn’t a cure. It’s a lease. And leases can expire.
Meina Taiwo
December 25, 2025 AT 17:35CF not contagious. Genetic. Need screening everywhere. Many die in Africa because no test. Simple fix: cheap DNA test kits in clinics. Done.
Southern NH Pagan Pride
December 26, 2025 AT 22:28Wait-so the government and Big Pharma teamed up to create a drug that fixes CF… but only for people who pass a genetic filter? And the rest are just… left to rot? Sounds like a controlled population experiment to me.
Did you know Vertex’s CEO used to work for the CIA? Coincidence? I think not.
Also, the ‘10% not helped’? That’s not a mutation problem. That’s a targeted deletion. They’re engineering who lives and who doesn’t. The sweat test? That’s a surveillance tool. You think they don’t track your chloride levels?
They’re not curing CF. They’re curating it.
Orlando Marquez Jr
December 27, 2025 AT 17:05The evolution of therapeutic intervention in cystic fibrosis represents a paradigmatic shift in the application of precision medicine to monogenic disorders. The pharmacodynamic efficacy of CFTR modulators, particularly in the context of F508del homozygosity, demonstrates statistically significant improvements in forced expiratory volume and reduction in pulmonary exacerbation frequency. However, the socioeconomic determinants of access remain a critical barrier to equitable health outcomes, particularly in low- and middle-income countries where diagnostic infrastructure remains under-resourced.
Further longitudinal studies are warranted to assess the long-term impact on organ system degeneration, particularly in pediatric cohorts exposed to modulators from infancy.
Jackie Be
December 28, 2025 AT 19:43TRIKAFTA GAVE ME MY LIFE BACK I WAS SO WEAK I COULDNT WALK UP THE STAIRS NOW I’M CLIMBING MOUNTAINS AND I JUST ATE A WHOLE PIZZA WITHOUT THROWING UP I CANT BELIEVE THIS IS REAL I THOUGHT I WAS GONNA DIE AT 25 AND NOW I’M PLANNING A TRIP TO JAPAN I LOVE SCIENCE I LOVE DOCTORS I LOVE EVERYONE WHO MADE THIS HAPPEN
TO THE 10% WHO STILL HURT-I SEE YOU. WE’RE FIGHTING FOR YOU TOO
John Hay
December 29, 2025 AT 16:55You’re all talking like this is some miracle. It’s not. It’s a drug. A damn expensive drug. And the people who need it most can’t get it. The fact that we’re celebrating a 50-year life expectancy like it’s a victory when it’s still half the national average is disgusting. This isn’t progress-it’s damage control. And we’re letting corporations decide who lives.
Stop pretending this is science. It’s profit with a heart emoji.